Docker Container Installation and Overview
We have provided a complete installation of the Immcantation framework, its dependencies, accessory scripts, and IgBLAST in a Docker container. The container also includes both the IgBLAST and IMGT reference germline sets, as well as several example pipeline scripts.
We currently have four containers available on DockerHub:
Name |
Contents |
|---|---|
immcantation/suite |
Immcantation suite, supporting applications and databases. |
immcantation/lab |
Immcantation tutorial materials. Only for training, not to be used in production. |
immcantation/base |
Base image for Immcantation builds. |
immcantation/test |
Immcantation unit test image. |
For tutorial purposes, use immcantation/lab (be sure to replace suite with lab in the following code chunks) and follow the directions here. For all Immcantation uses, use immcantation/suite.
Note that containers are versioned through tags with containers containing official releases
denoted by meta-version numbers (x.y.z). The devel tag denotes the
latest development (unstable) builds.
Getting the /suite Container
Requires an installation of Docker 1.9+ or Singularity 2.3+.
Docker
# Pull release version devel docker pull immcantation/suite:devel # Pull the latest development build docker pull immcantation/suite:devel
Our containers are Linux-based, so if you are using a Windows computer, please make sure that you are using Linux containers and not Windows containers (this can be changed in Docker Desktop and won’t affect your existing containers).
Singularity
# Pull release version devel IMAGE="immcantation_suite-devel.sif" singularity build $IMAGE docker://immcantation/suite:devel
The instructions to use containers from Docker Hub with Singularity can be slightly different for different versions of Singularity. If the command shown above doesn’t work for you, please visit Singularity Documentation and look for the specific command for your Singularity version under Build a container.
What’s in the /suite Container
Immcantation Tools
Third Party Tools
Example Pipeline Scripts
Accessory Scripts
The following accessory scripts are found in /usr/local/bin:
- fastq2fasta.py
Simple FASTQ to FASTA conversion.
- fetch_phix.sh
Downloads the PhiX174 reference genome.
- fetch_igblastdb.sh
Downloads the IgBLAST reference database.
- fetch_imgtdb.sh
Downloads the IMGT reference database.
- imgt2igblast.sh
Imports the IMGT reference database into IgBLAST.
- imgt2cellranger.py
Converts the IMGT fasta germline reference files to the input required by cellranger-mkvdjref.
Data
/usr/local/share/germlines/imgt/IMGT.yamlInformation about the downloaded IMGT reference sequences.
/usr/local/share/germlines/imgt/<species>/vdjDirectory containing IMGT-gapped V(D)J reference sequences in FASTA format.
/usr/local/share/igblastIgBLAST data directory.
/usr/local/share/igblast/fastaDirectory containing ungapped IMGT references sequences with IGH/IGK/IGL and TRA/TRB/TRG/TRD combined into single FASTA files, respectively.
/usr/local/share/protocolsDirectory containing primer, template switch and internal constant region sequences for various experimental protocols in FASTA format.
Using the Container
Invoking a shell inside the container
To invoke a shell session inside the container:
# Docker command docker run -it immcantation/suite:devel bash # Singularity command singularity shell immcantation_suite-devel.sif
Executing a specific command
After invoking an interactive session inside the container, commands can be executed in the container shell as they would be executed in the host shell.
Alternatively, it is possible to execute a specific command directly inside the
container without starting an interactive session. The next example demonstrates
how to execute ls within $HOME/project mounted to /data:
# Docker command docker run -v $HOME/project:/data:z immcantation/suite:devel ls /data # Singularity command singularity exec -B $HOME/project:/data immcantation_suite_|docker-version|.sif ls /data
Inspecting the container components
The container includes three informational scripts that provide details about the versions of installed software and available pipelines.
The
versions reportcommand will inspect the installed software versions and print them to standard output.The analogous
builds reportcommand will display the build date and changesets used during the image build. This is particularly relevant if you are using theimmcantation/suite:develdevelopment builds.Finally, the
pipelines reportcommand will display a list of available example pipelines included in the container.
Each command can be run using:
# Docker command docker run immcantation/suite:devel [command] # Singularity command singularity exec immcantation_suite_|docker-version|.sif [command]
Using the container for tutorials
If you would like to run and interact with the .Rmd tutorials from your
Docker container, run the following command (replace devel with a
release version if applicable):
docker run -it --rm -p 8787:8787 -e PASSWORD=immcantation immcantation/lab:devel
You can change the password to another of your choice. Note that in the container, RStudio uses the default port 8787.
Once the container is running, visit the url http://localhost:8787 to launch RStudio.
Use user magus and the password you provided in the previous command.
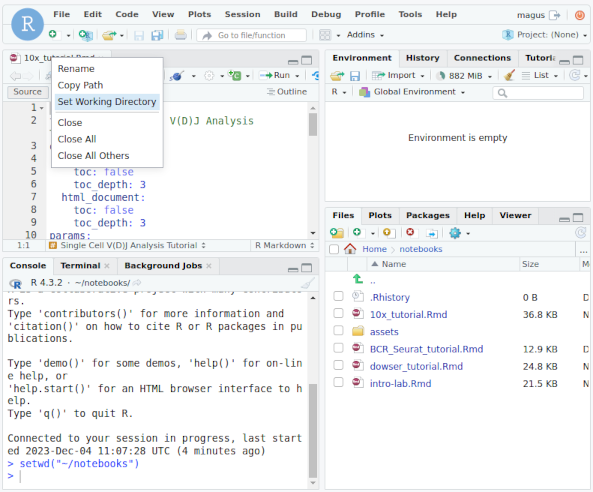
The folder notebooks contains .Rmd tutorials that can be executed in the container.
You can open one of the .Rmd files, set the working directory to ~notebooks, and knit
the tutorial or start running chunks.
Note: If you want to save the results locally in your computer, you need to bind the folder where you want to save the results to (<my-out-dir>), and the results folder in the container.
# change my-out-dir to the local directory where you want to have the results saved to docker run -it --rm \ -v <my-out-dir>:/home/magus/notebooks/results:z \ -p 8787:8787 immcantation/lab:devel
Example pipelines
You can always run your own pipeline scripts through the container, but the
container also includes a set of predefined pipeline scripts that can be run as
is or extended to your needs. Each pipeline script has a -h argument which
will explain its use. The available pipelines are:
preprocess-phix
presto-abseq
presto-clontech
presto-clontech-umi
changeo-10x
changeo-igblast
tigger-genotype
shazam-threshold
changeo-clone
All example pipeline scripts can be found in /usr/local/bin.
PhiX cleaning pipeline
Removes reads from a sequence file that align against the PhiX174 reference genome.
- Usage: preprocess-phix [OPTIONS]
- -s
FASTQ sequence file.
- -r
Directory containing phiX174 reference db. Defaults to /usr/local/share/phix.
- -n
Sample identifier which will be used as the output file prefix. Defaults to a truncated version of the input filename.
- -o
Output directory. Will be created if it does not exist. Defaults to a directory matching the sample identifier in the current working directory.
- -p
Number of subprocesses for multiprocessing tools. Defaults to the available cores.
- -h
This message.
Example: preprocess-phix
# Arguments
DATA_DIR=~/project
READS=/data/raw/sample.fastq
OUT_DIR=/data/presto/sample
NPROC=4
# Run pipeline in docker image
docker run -v $DATA_DIR:/data:z immcantation/suite:devel \
preprocess-phix -s $READS -o $OUT_DIR -p $NPROC
# Singularity command
singularity exec -B $DATA_DIR:/data immcantation_suite-devel.sif \
preprocess-phix -s $READS -o $OUT_DIR -p $NPROC
Note
The PhiX cleaning pipeline will convert the sequence headers to
the pRESTO format. Thus, if the nophix output file is provided as
input to the presto-abseq pipeline script you must pass the argument
-x presto to presto-abseq, which will tell the
script that the input headers are in pRESTO format (rather than the
Illumina format).
NEBNext / AbSeq immune sequencing kit preprocessing pipeline
A start to finish pRESTO processing script for NEBNext / AbSeq immune sequencing data.
An example for human BCR processing is shown below. Primer sequences are available from the
Immcantation repository under protocols/AbSeq
or inside the container under /usr/local/share/protocols/AbSeq. Mouse primers are not supplied.
TCR V gene references can be specified with the flag
-r /usr/local/share/igblast/fasta/imgt_human_tr_v.fasta.
- Usage: presto-abseq [OPTIONS]
- -1
Read 1 FASTQ sequence file. Sequence beginning with the C-region or J-segment).
- -2
Read 2 FASTQ sequence file. Sequence beginning with the leader or V-segment).
- -j
Read 1 FASTA primer sequences. Defaults to /usr/local/share/protocols/AbSeq/AbSeq_R1_Human_IG_Primers.fasta.
- -v
Read 2 FASTA primer or template switch sequences. Defaults to /usr/local/share/protocols/AbSeq/AbSeq_R2_TS.fasta.
- -c
C-region FASTA sequences for the C-region internal to the primer. If unspecified internal C-region alignment is not performed.
- -r
V-segment reference file. Defaults to /usr/local/share/igblast/fasta/imgt_human_ig_v.fasta.
- -y
YAML file providing description fields for report generation.
- -n
Sample identifier which will be used as the output file prefix. Defaults to a truncated version of the read 1 filename.
- -o
Output directory. Will be created if it does not exist. Defaults to a directory matching the sample identifier in the current working directory.
- -x
The mate-pair coordinate format of the raw data. Defaults to illumina.
- -p
Number of subprocesses for multiprocessing tools. Defaults to the available cores.
- -h
This message.
One of the requirements for generating the report at the end of the pRESTO pipeline is a YAML
file containing information about the data and processing. Valid fields are shown in the example
sample.yaml below, although no fields are strictly required:
sample.yaml
title: "pRESTO Report: CD27+ B cells from subject HD1"
author: "Your Name"
version: "0.5.4"
description: "Memory B cells (CD27+)."
sample: "HD1"
run: "ABC123"
date: "Today"
Example: presto-abseq
# Arguments
DATA_DIR=~/project
READS_R1=/data/raw/sample_R1.fastq
READS_R2=/data/raw/sample_R2.fastq
YAML=/data/sample.yaml
SAMPLE_NAME=sample
OUT_DIR=/data/presto/sample
NPROC=4
# Docker command
docker run -v $DATA_DIR:/data:z immcantation/suite:devel \
presto-abseq -1 $READS_R1 -2 $READS_R2 -y $YAML \
-n $SAMPLE_NAME -o $OUT_DIR -p $NPROC
# Singularity command
singularity exec -B $DATA_DIR:/data immcantation_suite-devel.sif \
presto-abseq -1 $READS_R1 -2 $READS_R2 -y $YAML \
-n $SAMPLE_NAME -o $OUT_DIR -p $NPROC
Takara Bio / Clontech SMARTer v1 immune sequencing kit preprocessing pipeline
A start to finish pRESTO processing script for Takara Bio / Clontech SMARTer v1 immune
sequencing kit data. C-regions are assigned using the universal C-region primer sequences are
available from the Immcantation repository under
protocols/Universal
or inside the container under /usr/local/share/protocols/Universal.
- Usage: presto-clontech [OPTIONS]
- -1
Read 1 FASTQ sequence file. Sequence beginning with the C-region.
- -2
Read 2 FASTQ sequence file. Sequence beginning with the leader.
- -j
C-region reference sequences (reverse complemented). Defaults to /usr/local/share/protocols/Universal/Mouse_IG_CRegion_RC.fasta.
- -r
V-segment reference file. Defaults to /usr/local/share/igblast/fasta/imgt_mouse_ig_v.fasta.
- -y
YAML file providing description fields for report generation.
- -n
Sample identifier which will be used as the output file prefix. Defaults to a truncated version of the read 1 filename.
- -o
Output directory. Will be created if it does not exist. Defaults to a directory matching the sample identifier in the current working directory.
- -x
The mate-pair coordinate format of the raw data. Defaults to illumina.
- -p
Number of subprocesses for multiprocessing tools. Defaults to the available cores.
- -h
This message.
Example: presto-clontech
# Arguments
DATA_DIR=~/project
READS_R1=/data/raw/sample_R1.fastq
READS_R2=/data/raw/sample_R2.fastq
CREGION=/usr/local/share/protocols/Universal/Human_IG_CRegion_RC.fasta
VREF=/usr/local/share/igblast/fasta/imgt_human_ig_v.fasta
SAMPLE_NAME=sample
OUT_DIR=/data/presto/sample
NPROC=4
# Docker command
docker run -v $DATA_DIR:/data:z immcantation/suite:devel \
presto-clontech -1 $READS_R1 -2 $READS_R2 -j $CREGION -r $VREF \
-n $SAMPLE_NAME -o $OUT_DIR -p $NPROC
# Singularity command
singularity exec -B $DATA_DIR:/data immcantation_suite-devel.sif \
presto-clontech -1 $READS_R1 -2 $READS_R2 -j $CREGION -r $VREF \
-n $SAMPLE_NAME -o $OUT_DIR -p $NPROC
Takara Bio / Clontech SMARTer v2 (UMI) immune sequencing kit preprocessing pipeline
A start to finish pRESTO processing script for Takara Bio / Clontech SMARTer v2 immune
sequencing kit data that includes UMIs. C-regions are assigned using the universal C-region
primer sequences are available from the Immcantation repository under
protocols/Universal
or inside the container under /usr/local/share/protocols/Universal.
- Usage: presto-clontech-umi [OPTIONS]
- -1
Read 1 FASTQ sequence file. Sequence beginning with the C-region.
- -2
Read 2 FASTQ sequence file. Sequence beginning with the leader.
- -j
C-region reference sequences (reverse complemented). Defaults to /usr/local/share/protocols/Universal/Human_IG_CRegion_RC.fasta.
- -r
V-segment reference file. Defaults to /usr/local/share/igblast/fasta/imgt_human_ig_v.fasta.
- -n
Sample identifier which will be used as the output file prefix. Defaults to a truncated version of the read 1 filename.
- -o
Output directory. Will be created if it does not exist. Defaults to a directory matching the sample identifier in the current working directory.
- -x
The mate-pair coordinate format of the raw data. Defaults to illumina.
- -p
Number of subprocesses for multiprocessing tools. Defaults to the available cores.
- -a
Specify to run multiple alignment of barcode groups prior to consensus. This step is skipped by default.
- -h
This message.
Example: presto-clontech-umi
# Arguments
DATA_DIR=~/project
READS_R1=/data/raw/sample_R1.fastq
READS_R2=/data/raw/sample_R2.fastq
CREGION=/usr/local/share/protocols/Universal/Human_IG_CRegion_RC.fasta
VREF=/usr/local/share/igblast/fasta/imgt_human_ig_v.fasta
SAMPLE_NAME=sample
OUT_DIR=/data/presto/sample
NPROC=4
# Docker command
docker run -v $DATA_DIR:/data:z immcantation/suite:devel \
presto-clontech-umi -1 $READS_R1 -2 $READS_R2 -j $CREGION -r $VREF \
-n $SAMPLE_NAME -o $OUT_DIR -p $NPROC
# Singularity command
singularity exec -B $DATA_DIR:/data immcantation_suite-devel.sif \
presto-clontech-umi -1 $READS_R1 -2 $READS_R2 -j $CREGION -r $VREF \
-n $SAMPLE_NAME -o $OUT_DIR -p $NPROC
10x Genomics V(D)J annotation pipeline
Assigns new annotations and infers clonal relationships to 10x Genomics single cell V(D)J data output by Cell Ranger.
- Usage: changeo-10x [OPTIONS]
- -s
FASTA or FASTQ sequence file.
- -a
10x Genomics cellranger-vdj contig annotation CSV file. Must corresponding with the FASTA/FASTQ input file (all, filtered or consensus).
- -r
Directory containing IMGT-gapped reference germlines. Defaults to /usr/local/share/germlines/imgt/[species name]/vdj.
- -g
Species name. One of human, mouse, rabbit, rat, or rhesus_monkey. Defaults to human.
- -t
Receptor type. One of ig or tr. Defaults to ig.
- -x
Distance threshold for clonal assignment. Specify “auto” for automatic detection. If unspecified, clonal assignment is not performed.
- -m
Distance model for clonal assignment. Defaults to the nucleotide Hamming distance model (ham).
- -e
Method to use for determining the optimal threshold. One of ‘gmm’ or ‘density’. Defaults to ‘density’.
- -d
Curve fitting model. Applies only when method (-e) is ‘gmm’. One of ‘norm-norm’, ‘norm-gamma’, ‘gamma-norm’ and ‘gamma-gamma’. Defaults to ‘gamma-gamma’.
- -u
Method to use for threshold selection. Applies only when method (-e) is ‘gmm’. One of ‘optimal’, ‘intersect’ and ‘user’. Defaults to ‘user’.
- -b
IgBLAST IGDATA directory, which contains the IgBLAST database, optional_file and auxillary_data directories. Defaults to /usr/local/share/igblast.
- -n
Sample identifier which will be used as the output file prefix. Defaults to a truncated version of the sequence filename.
- -o
Output directory. Will be created if it does not exist. Defaults to a directory matching the sample identifier in the current working directory.
- -f
Output format. One of changeo or airr. Defaults to airr.
- -p
Number of subprocesses for multiprocessing tools. Defaults to the available cores.
- -i
Specify to allow partial alignments.
- -z
Specify to disable cleaning and compression of temporary files.
- -h
This message.
Example: changeo-10x
# Arguments
DATA_DIR=~/project
READS=/data/raw/sample_filtered_contig.fasta
ANNOTATIONS=/data/raw/sample_filtered_contig_annotations.csv
SAMPLE_NAME=sample
OUT_DIR=/data/changeo/sample
DIST=auto
NPROC=4
# Run pipeline in docker image
docker run -v $DATA_DIR:/data:z immcantation/suite:devel \
changeo-10x -s $READS -a $ANNOTATIONS -x $DIST -n $SAMPLE_NAME \
-o $OUT_DIR -p $NPROC
# Singularity command
singularity exec -B $DATA_DIR:/data immcantation_suite-devel.sif \
changeo-10x -s $READS -a $ANNOTATIONS -x $DIST -n $SAMPLE_NAME \
-o $OUT_DIR -p $NPROC
IgBLAST annotation pipeline
Performs V(D)J alignment using IgBLAST and post-processes the output into the Change-O data standard.
- Usage: changeo-igblast [OPTIONS]
- -s
FASTA or FASTQ sequence file.
- -r
Directory containing IMGT-gapped reference germlines. Defaults to /usr/local/share/germlines/imgt/[species name]/vdj.
- -g
Species name. One of human, mouse, rabbit, rat, or rhesus_monkey. Defaults to human.
- -t
Receptor type. One of ig or tr. Defaults to ig.
- -b
IgBLAST IGDATA directory, which contains the IgBLAST database, optional_file and auxillary_data directories. Defaults to /usr/local/share/igblast.
- -n
Sample identifier which will be used as the output file prefix. Defaults to a truncated version of the sequence filename.
- -o
Output directory. Will be created if it does not exist. Defaults to a directory matching the sample identifier in the current working directory.
- -f
Output format. One of airr (default) or changeo. Defaults to airr.
- -p
Number of subprocesses for multiprocessing tools. Defaults to the available cores.
- -k
Specify to filter the output to only productive/functional sequences.
- -i
Specify to allow partial alignments.
- -z
Specify to disable cleaning and compression of temporary files.
- -h
This message.
Example: changeo-igblast
# Arguments
DATA_DIR=~/project
READS=/data/presto/sample/sample-final_collapse-unique_atleast-2.fastq
SAMPLE_NAME=sample
OUT_DIR=/data/changeo/sample
NPROC=4
# Run pipeline in docker image
docker run -v $DATA_DIR:/data:z immcantation/suite:devel \
changeo-igblast -s $READS -n $SAMPLE_NAME -o $OUT_DIR -p $NPROC
# Singularity command
singularity exec -B $DATA_DIR:/data immcantation_suite-devel.sif \
changeo-igblast -s $READS -n $SAMPLE_NAME -o $OUT_DIR -p $NPROC
Genotyping pipeline
Infers V segment genotypes using TIgGER.
- Usage: tigger-genotype [options]
- -d DB, --db=DB
Change-O formatted TSV (TAB) file.
- -r REF, --ref=REF
FASTA file containing IMGT-gapped V segment reference germlines. Defaults to /usr/local/share/germlines/imgt/human/vdj/imgt_human_IGHV.fasta.
- -v VFIELD, --vfield=VFIELD
Name of the output field containing genotyped V assignments. Defaults to V_CALL_GENOTYPED.
- -x MINSEQ, --minseq=MINSEQ
Minimum number of sequences in the mutation/coordinate range. Samples with insufficient sequences will be excluded. Defaults to 50.
- -y MINGERM, --mingerm=MINGERM
Minimum number of sequences required to analyze a germline allele. Defaults to 200.
- -n NAME, --name=NAME
Sample name or run identifier which will be used as the output file prefix. Defaults to a truncated version of the input filename.
- -u FIND-UNMUTATED, --find-unmutated=FIND-UNMUTATED
Whether to use ‘-r’ to find which samples are unmutated. Defaults to TRUE.
- -o OUTDIR, --outdir=OUTDIR
Output directory. Will be created if it does not exist. Defaults to the current working directory.
- -f FORMAT, --format=FORMAT
File format. One of ‘airr’ (default) or ‘changeo’.
- -p NPROC, --nproc=NPROC
Number of subprocesses for multiprocessing tools. Defaults to the available processing units.
- -h, --help
Show this help message and exit
Example: tigger-genotype
# Arguments
DATA_DIR=~/project
DB=/data/changeo/sample/sample_db-pass.tab
SAMPLE_NAME=sample
OUT_DIR=/data/changeo/sample
NPROC=4
# Run pipeline in docker image
docker run -v $DATA_DIR:/data:z immcantation/suite:devel \
tigger-genotype -d $DB -n $SAMPLE_NAME -o $OUT_DIR -p $NPROC
# Singularity command
singularity exec -B $DATA_DIR:/data immcantation_suite-devel.sif \
tigger-genotype -d $DB -n $SAMPLE_NAME -o $OUT_DIR -p $NPROC
TIgGER infers the subject-specific genotyped V gene calls and saves the corrected calls in a new column, v_call_genotyped.
TIgGER also generates a *_genotype.fasta file, which contains the subject-specific germline IGHV genes. In future analyses,
if v_call_genotyped column is used to replace v_call, please remember to use this *_genotype.fasta file generated previously
by TIgGER as the subject-specific IGHV gene germline. An example of this application can be found in the Clonal assignment pipeline section.
Clonal threshold inference pipeline
Performs automated detection of the clonal assignment threshold.
- Usage: shazam-threshold [options]
- -d DB, --db=DB
Tabulated data file, in Change-O (TAB) or AIRR format (TSV).
- -m METHOD, --method=METHOD
Threshold inferrence to use. One of gmm, density, or none. If none, the distance-to-nearest distribution is plotted without threshold detection. Defaults to density.
- -n NAME, --name=NAME
Sample name or run identifier which will be used as the output file prefix. Defaults to a truncated version of the input filename.
- -o OUTDIR, --outdir=OUTDIR
Output directory. Will be created if it does not exist. Defaults to the current working directory.
- -f FORMAT, --format=FORMAT
File format. One of ‘airr’ (default) or ‘changeo’.
- -p NPROC, --nproc=NPROC
Number of subprocesses for multiprocessing tools. Defaults to the available processing units.
- --model=MODEL
Model to use for the gmm model. One of gamma-gamma, gamma-norm, norm-norm or norm-gamma. Defaults to gamma-gamma.
- --cutoff=CUTOFF
Method to use for threshold selection. One of optimal, intersect or user. Defaults to optimal.
- --spc=SPC
Specificity required for threshold selection. Applies only when method=’gmm’ and cutoff=’user’. Defaults to 0.995.
- --subsample=SUBSAMPLE
Number of distances to downsample the data to before threshold calculation. By default, subsampling is not performed.
- --repeats=REPEATS
Number of times to recalculate. Defaults to 1.
- -h, --help
Show this help message and exit
Example: shazam-threshold
# Arguments
DATA_DIR=~/project
DB=/data/changeo/sample/sample_genotyped.tab
SAMPLE_NAME=sample
OUT_DIR=/data/changeo/sample
NPROC=4
# Run pipeline in docker image
docker run -v $DATA_DIR:/data:z immcantation/suite:devel \
shazam-threshold -d $DB -n $SAMPLE_NAME -o $OUT_DIR -p $NPROC
# Singularity command
singularity exec -B $DATA_DIR:/data immcantation_suite-devel.sif \
shazam-threshold -d $DB -n $SAMPLE_NAME -o $OUT_DIR -p $NPROC
Clonal assignment pipeline
Assigns Ig sequences into clonally related lineages and builds full germline sequences.
If the TIgGER, or another package, was applied previously to the data set for
identifying a subject-specific genotype, including potentially novel V, D
and/or J genes, a new directory $NEW_REF with the personalized germline database
should be created. For example, if TIgGER was run to identify a subject-specific
IGHV genotype, the directory would contain: 1) *_genotype.fasta file generated
previously by TIgGER, which contains the subject-specific germline IGHV genes
2) imgt_human_IGHD.fasta and imgt_human _IGHJ.fasta, which contain the IMGT IGHD
and IGHJ genes and can both be copied from the original germline
database: /usr/local/share/germlines/imgt/human/vdj/. When changeo-clone is called,
this new personalized germline database should be passed with parameter -r
(see example below). And please remember to update v_call column with
subject-specific IGHV call (for TIgGER this is found in v_call_genotyped column).
# update v_call
db %>%
dplyr::mutate(v_call = v_call_genotyped) %>%
select(-v_call_genotyped)
- Usage: changeo-clone [OPTIONS]
- -d
Change-O formatted TSV (TAB) file.
- -x
Distance threshold for clonal assignment.
- -m
Distance model for clonal assignment. Defaults to the nucleotide Hamming distance model (ham).
- -r
Directory containing IMGT-gapped reference germlines. Defaults to /usr/local/share/germlines/imgt/human/vdj.
- -n
Sample identifier which will be used as the output file prefix. Defaults to a truncated version of the input filename.
- -o
Output directory. Will be created if it does not exist. Defaults to a directory matching the sample identifier in the current working directory.
- -f
Output format. One of airr (default) or changeo.
- -p
Number of subprocesses for multiprocessing tools. Defaults to the available cores.
- -a
Specify to clone the full data set. By default the data will be filtering to only productive/functional sequences.
- -z
Specify to disable cleaning and compression of temporary files.
- -h
This message.
Example: changeo-clone
# Arguments
DATA_DIR=~/project
DB=/data/changeo/sample/sample_genotyped.tab
DIST=0.15
SAMPLE_NAME=sample
OUT_DIR=/data/changeo/sample
NPROC=4
# Run pipeline in docker image
docker run -v $DATA_DIR:/data:z immcantation/suite:devel \
changeo-clone -d $DB -x $DIST -n $SAMPLE_NAME -o $OUT_DIR -p $NPROC
# Singularity command
singularity exec -B $DATA_DIR:/data immcantation_suite-devel.sif \
changeo-clone -d $DB -x $DIST -n $SAMPLE_NAME -o $OUT_DIR -p $NPROC
Example: changeo-clone with personalized germline database
# Arguments DATA_DIR=~/project NEW_REF=/data/personalized_germlines DB=/data/changeo/sample/sample_genotyped.tab DIST=0.15 SAMPLE_NAME=sample OUT_DIR=/data/changeo/sample NPROC=4 # Run pipeline in docker image docker run -v $DATA_DIR:/data:z immcantation/suite:devel changeo-clone -r $NEW_REF -d $DB -x $DIST -n $SAMPLE_NAME -o $OUT_DIR -p $NPROC # Singularity command singularity exec -B $DATA_DIR:/data immcantation_suite-devel.sif changeo-clone -r $NEW_REF -d $DB -x $DIST -n $SAMPLE_NAME -o $OUT_DIR -p $NPROC